See full prescribing information for AIKENINGTM and use it under the instruction of medical professionals.
AIKENINGTM(Albuvirtide for injection) for intravenous use
Initial China Approval: May 23, 2018
[Product Name]
Generic name: Albuvirtide for injection
[Ingredients]
Active pharmaceutical ingredient (API): Albuvirtide
Chemical name:
Acetyl-Trp-Glu-Glu-Trp-Asp-Arg-Glu-Ile-Asn-Asn-Tyr-Thr-(N-{2-[2-(N-(3-maleimidepropionyl) amino) ethoxy] ethoxy}acetyl) Lys-Leu-Ile-His-Glu-Leu-Ile-Glu-Glu- Ser-Gln-Asn-Gln-Gln-Glu-Lys-Asn-Glu-Gln-Glu-Leu-Leucinamide
Chemical structure:
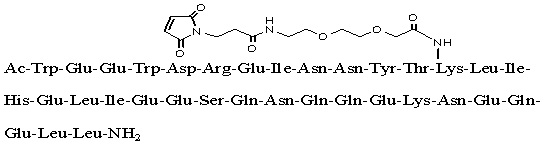
Molecular formula: C204H306N54O72
Molecular weight: 4666.93
This product contains no pharmaceutical excipients.
[Description]
Off-white or yellowish porous cake or powder.
[Indications]
Albuvirtide is a human immunodeficiency virus type 1 (HIV-1) fusion inhibitor indicated in combination with other antiretroviral agent(s) for treatment of HIV-1 infection in treatment-experienced patients with HIV-1 replication despite ongoing antiretroviral therapy.
[Strength]
160 mg/vial.
[Dosage and Administration]
1. Dosage regimen
For adults and adolescents over 16 years old, AIKENINGTM is administered by intravenous infusion at a dose of 320 mg once a day on Day 1, 2, 3, and 8, and thereafter once a week.
2. Preparation of AIKENINGTM

Step 1: Determine number of AIKENINGTM vial(s) needed for infusion based on assigned dosage (see table above). For 0.32 g dose, two vials are needed.
· Have all the materials ready before starting the dose preparation to minimize the overall preparation time. Use aseptic technique when preparing and administering the dose.
· From a 100 mL infusion bag (bottle) of 0.9% sodium chloride solution withdraw 12 mL to discard. The remaining 88 mL of 0.9% sodium chloride solution will be used for the AIKENINGTM reconstitution.
Step 2: Inject 1.2 mL of 5% sodium bicarbonate solution into each vial, shake or vortex the vial thoroughly to form solution.
· When preparing for two vials of AIKENINGTM, do not withdraw 2.4 mL of sodium bicarbonate solution in one syringe and attempt to split the volume into two vials.
· Note: It will be difficult to control the volume of sodium bicarbonate solution injected into the vial as the vials are sealed under vacuum (negative air pressure) which will likely create a suction.
· Upon addition of sodium bicarbonate, immediately vibrate the vial (within 30 seconds) using a vortex mixer at a setting of 2500 rpm to dissolve AIKENINGTM powder into the sodium bicarbonate solution.
· The complete dissolution is normally achieved within 5 min; if undissolved solid is observed after 5 min of mixing, continue using a vortex mixer or manually shaking till a complete dissolution. Discard the vial if a complete dissolution was not achieved in 20 min of mixing; re-prepare the dose with a fresh vial.
Step 3: Withdraw 6 mL of sodium chloride solution from remaining 88 mL of 0.9% sodium chloride solution in Step 1 and inject into each AIKENINGTM vial to mix thoroughly.
· Gently mix AIKENINGTM solution in sodium bicarbonate with sodium chloride solution in the vial to avoid excessive bubbling. The resulted solution should be clear, transparent, and free of visible particulate.
Step 4: Withdraw entire contents of vial(s) and inject back into the original bag (bottle) of the remaining 0.9% sodium chloride solution in Step 3 to get a final total of infusion volume of ~90 mL AIKENINGTM solution.
· The prepared dose should be administered as soon as possible (or within 30 minutes after end of reconstitution). Discard the dose if the infusion is not started within 30 min after the end of dose preparation and re-prepare the dose with a fresh vial.
· The storage of reconstituted drug product in refrigerator or freezer is not allowed.
3. Administration
Administer AIKENINGTM solution by intravenous infusion via a peripheral vein in one of the upper extremities for 45±8 minutes.
[Adverse Reactions]
Two Phase I, one Phase II clinical trials of AIKENINGTM have been completed, and one Phase III is ongoing and an interim analysis is reported. A total of 294 infected subjects were enrolled in the trials. 170 patients with HIV-1 infection have been exposed to AIKENINGTM. The safety assessment of AIKENINGTM was primarily based on the ongoing randomized, controlled phase III clinical trial (TALENT study) in HIV-1 infected patients previously treated with antiretrovirals, in which the treatment group received the AIKENINGTM (once a week, intravenous infusion) and Lopinavir/ Ritonavir (LPV/r) combination therapy, and the control group received the standard 3-drug regimen of LPV/r + Tenofovir (TDF) or Zidovudine (AZT) + Lamivudine (3TC). The interim analysis summarized the safety data of the treatment group (n = 93) and the control group (n = 99) patients who completed 24 to 48-week treatment.
Adverse reactions
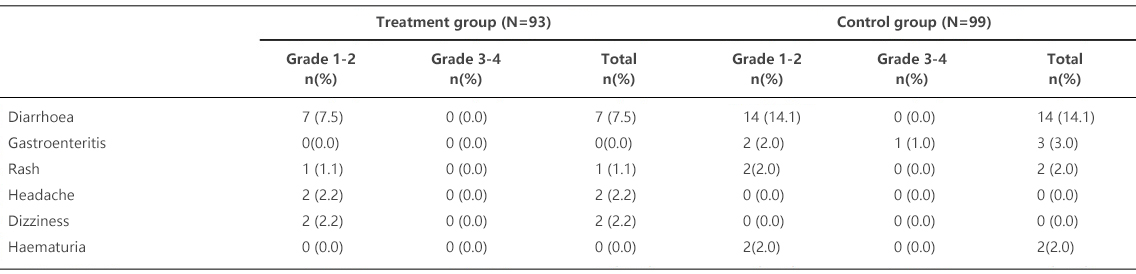
Table 1 summarizes clinical adverse reactions with incidence rates ≥ 2%. The most commonly reported adverse reactions include diarrhea, headache, dizziness and rash.
Table 1. Adverse Reactions with Incidence Rates ≥ 2%
Laboratory abnormalities
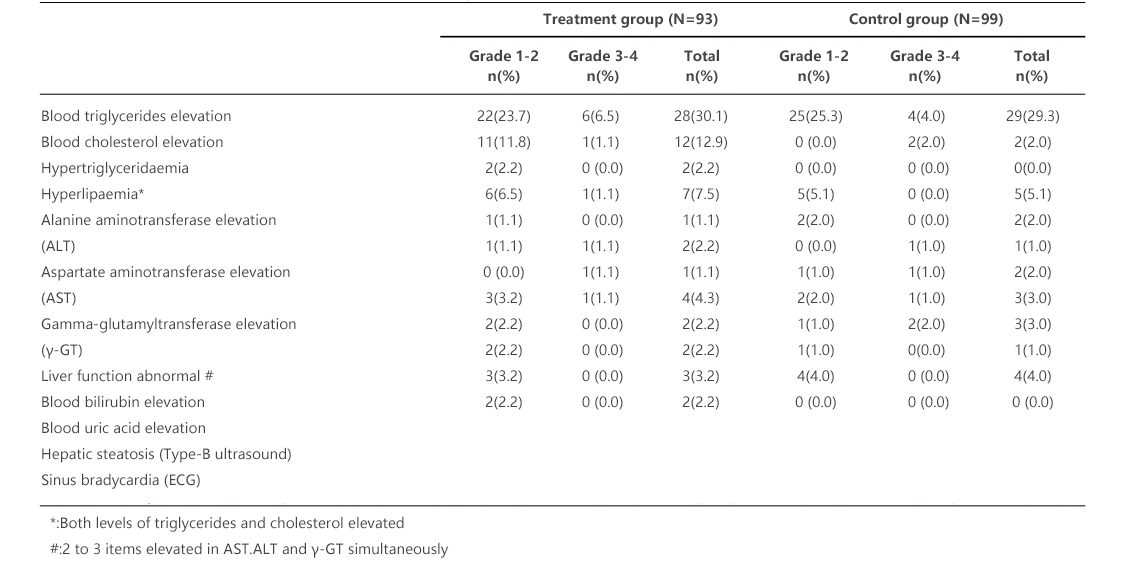
Table 2 summarizes drug-related abnormal laboratory values with incidence rates ≥2%. The commonly reported abnormal laboratory values were elevated level of triglycerides and hyperlipidemia, such as hyperlipaemia, hypertriglyceridaemia, elevated alanine transaminase (ALT) level, elevated aspartate aminotransferase (AST) level, elevated Gamma-glutamyltransferase (γ-GT) level, bilirubin and serum uric acid, etc. Most of them were mild to moderate elevations (grade 1 - grade 2). The incidence rate of elevated total cholesterol was higher in the treatment group than that in the control group. In addition, there was no statistically significant difference between the two groups.
Table 2. Laboratory Abnormalities with Incidence Rates ≥2%
[Contraindications]
This product is contraindicated in patients allergic to its ingredients.
[Precautions]
1. This product should be clear, transparent solution after being dissolved, and must not be used if turbidity, precipitate or foreign substances are observed.
2. The reconstituted solution should be infused for once, and it should not be used separately.
[Pregnancy and breastfeeding]
In the reproductive toxicity studies in rat and rabbit, the No Observed Adverse Effect Level (NOAEL) of Albuvirtide intravenously injected were 4 folds and 2 folds of human equivalent dose, respectively, and no toxicity to parental fertility and embryonic development was observed. Since no clinical data are available in pregnant women, it is not recommended that this product be used for pregnant women.
It is unknown whether Albuvirtide is excreted in human breast milk. Mothers with HIV-infections should not breast-feed infants so as to avoid HIV transmission. Mothers should not breastfeed infants if they are receiving AIKENINGTM.
[Children]
The safety and efficacy of AIKENINGTM have not been established in children under 16 years old.
[Geriatric]
No adequate human data are available in geriatric patients aged over 65 years; it is unknown whether their response to AIKENINGTM is different from young people.
[Drug Interactions]
In the in vitro human liver microsome test, Albuvirtide was demonstrated not a CYP450 enzyme inhibitor, and had no significant inhibition effect on the activity of human liver microsomal enzymes CYP1A2, 2C8, 2C9, 2C19, 2D6 and 3A4.
In the in vitro anti-HIV-1 combination therapy, Albuvirtide had synergistic effect with zidovudine (AZT) and saquinavir (SQV), and additive effect with efavirenz (EFV) and enfuvirtide (T20).
Combination of AIKENINGTM and lopinavir/ritonavir (LPV/r) did not change the pharmacokinetics profile of Albuvirtide, the in vivo exposure to LPV/r was reduced, and no dose adjustments are needed.
[Overdose]
At present there is no information on overdose of AIKENINGTM in humans. Six subjects with HIV-1 infections in clinical trials received single intravenous infusion at highest dose of 640 mg, but no drug-related adverse reaction was observed. There is currently no specific antidote against overdose of AIKENINGTM.
[Clinical Trials]
The efficacy of AIKENINGTM is mainly based on analyses of data from a dose exploration study and an on-going pivotal study (TALENT study).
The dose-exploration study was an open, parallel study designed to evaluate the efficacy and safety of combination AIKENINGTM and lopinavir/ritonavir (LPV/r) for the treatment of HIV-1 infection. Twenty treatment-naive HIV-1 infected patients were randomized and received AIKENINGTM 160 mg or 320 mg once a week, both combination with lopinavir/ritonavir. The primary efficacy endpoint was the mean change of HIV-1 RNA from baseline at the end of treatment. The results showed that after treatment with combination of AIKENINGTM and LPV/r, HIV-RNA decreased significantly in all 20 subjects, and the CD4 cell count increased from baseline to different extents. The antiviral effect of AIKENINGTM 320 mg group was significantly better than that of the 160 mg group.
TALENT was a multicenter, open, randomized, non-inferiority clinical phase III study. The objective was to evaluate the safety and efficacy of AIKENINGTM combined with LPV/r for treatment of HIV-1 infected patients who failed standard first-line ART. The study was designed to enroll 420 subjects for 48-week treatment with 7 visits. All subjects were randomized at 1:1 ratio to the treatment and control group. Subjects in the treatment group received AIKENINGTM 320 mg once a day on day 1, 2, 3 and 8, and then once a week, combined with LPV/r for 48 weeks. Subjects in the control group received a 3-drug combination of LPV/r + Tenofovir (TDF) or Zidovudine (AZT) + Lamivudine (3TC) for 48 weeks. The primary efficacy endpoint was the proportion of patients with the HIV RNA < 50 copies/ml at 48 week (by FDA Snapshot Algorithm). The second efficacy endpoints included the mean change of HIV RNA from baseline, the proportion of patients with the HIV RNA < 400 copies/ml (by FDA Snapshot Algorithm), and the mean change of CD4 cell count from baseline after treatment.
Interim analysis summarized the efficacy data for the treatment group (n = 83) and the control group (n = 92) in which subjected were treated for 24 to 48 weeks.
The percentages of patients with HIV-RNA <50 copies/ml after treatment for 48 weeks were 80.4% and 66.0%, respectively in the treatment group and the control group in the FAS analysis set, and the bilateral 95% CI of treatment difference between the two groups was -3.0% ~ 31.9%, meeting the criterion for non-inferiority with a prespecified margin of 12%. It indicated that efficacy of the treatment group was non-inferior to that of the control group (see Table 3). The percentages of subjects with HIV-RNA <50 copies/ml were 94.9% and 74.4%, respectively (P <0.05, with statistical significance) in the treatment group and the control group in the PPS analysis set, indicating that the efficacy in the treatment group was superior to that of the control group.
Logistic regression analysis showed that gender, baseline viral load and CD4 cell count had no significant effect on the percentage of patients with HIV-RNA <50 copies/ml at Week 48, and did not affect the therapeutic effect. Albuvirtide could effectively suppress virus replication in HIV-infected subjects with a viral load over 100,000 copies/ml and those with CD4 cell count below 100 cells/μl (see Table 3).
Table 3. Primary and Secondary Efficacy Endpoints (FAS Analysis Set)
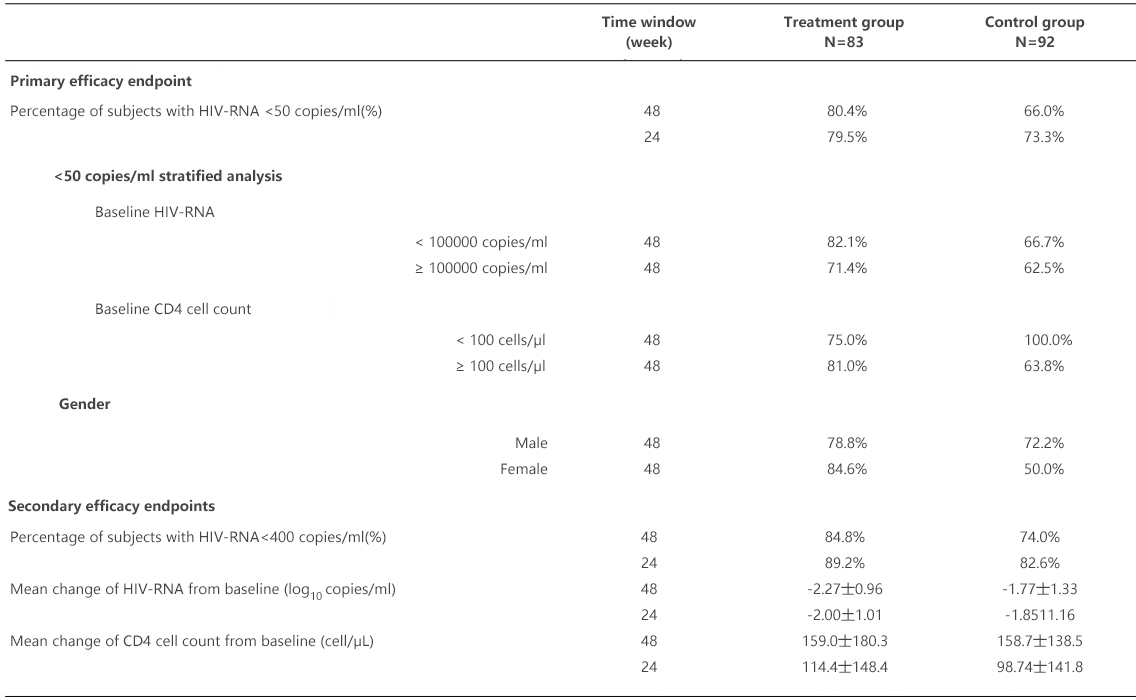
The percentages of subjects with the primary efficacy endpoint of HIV-RNA <50 copies/ml after treatment for 24 weeks were 79.5% and 78.3%, respectively in the treatment group and the control group in the FAS analysis set, and the bilateral 95% CI of treatment difference between the two groups was -10.8% ~ 13.4%, meeting the criterion for non-inferiority with a prespecified margin of 12%, but without statistically significant difference (P>0.05). It indicated that efficacy of the treatment group was non-inferior to that of the control group. The result of the PPS analysis consistent with the FAS analysis.
The secondary efficacy endpoints included the mean change of HIV-RNA from baseline (log10 copies/ml), percentage of subjects with HIV-RNA < 400 copies/ml after treatment and mean change of CD4 cell count from baseline (cell/μL), all of them have basically consistent results with the primary efficacy endpoint (see Table 3).
[Pharmacology and Toxicology]
Pharmacology
Mechanism of action:
Albuvirtide is an HIV fusion inhibitor targeting the HIV-1 viral envelope subunit glycoprotein 41 (gp41). It inhibits the fusion of viruses with human target cells, thus blocking viruses from entering cells.
Antiviral activity:
In vitro antiretroviral activity Albuvirtide-albumin conjugates against various HIV-1 strains showed the IC50 of Albuvirtide against 8 HIV-1 subtypes (A, B, C, G and EA recombinants) was 0.5 ~ 4.8 nM in human peripheral blood mononuclear cells (PBMCs). The mean IC50 of Albuvirtide against 28 prevalent strains of CRF07-BC, CRF01-AE, and B' subtypes from China were 5.2 nM, 6.9 nM and 9.5 nM, respectively.
The in vivo antiviral activity of subcutaneous injection of Albuvirtide was evaluated in SCID-hu Thy/Liv mice model that was constructed by transplantation of human fetal thymus and liver into immunodeficient CB-17-SCID mice and inoculation with HIV-1. Albuvirtide showed strong antiviral activity in mice at a dose of 10 mg/kg whether Albuvirtide was administered by subcutaneous injection once a day or every other day (equal to around 2 and 4 half-life intervals of mouse albumin, respectively).
Drug resistance:
In vitro selection of resistant viruses to Albuvirtide showed that Albuvirtide showed a high resistance gene barrier. The resistant virus was achieved by 9 passages of HIV-1 in the presence of Albuvirtide with a sensitivity decrease of 159 folds, and the resistant viruses were across resistant to T20. The main mutations were Q40K, N126K and K114I.
The interim analysis of Albuvirtide phase III trial showed that no resistant mutations associated with the fusion inhibitor were found in HIV-1 gp41 from 5 patients who had HIV-RNA > 400 copies/ml after combination therapy of Albuvirtide and LPV/r for 24 to 48 weeks.
Cross-resistance:
In vitro assay showed that seven laboratory HIV-1 strains resistant to T20 with resistant mutation in the 36, 38, 42 and 43 site were sensitive to Albuvirtide.
Toxicology
Genetic toxicity:
The genetic toxicity tests of Albuvirtide are negative, including the bacterial reverse mutation test (Ames test), in vitro chromosome aberration test of CHL cells and micronucleus test in bone marrow of mice.
Reproductive toxicity:
In the fertility and early embryo development toxicity study in Wistar rats, Albuvirtide was administered by intravenous injection at 30, 60 and 120 mg/kg. Male rats of each group were treated from 4 weeks before mating to the dissection, and females were treated from two weeks before mating to the eighth day of pregnancy. The frequency of administration was once every 2 days. In all Albuvirtide dose groups; there was no interference or toxic effect on the fertility and early embryo development in male and female rats. The NOAEL of Albuvirtide for the reproductive function, formation and development of embryos in the parental rats was determined to be 120 mg/kg, i.e. 4 folds of human equivalent dose calculated in terms of body surface area.
In the embryo-fetal development toxicity study in Wistar rats, the pregnant rats were given Albuvirtide by intravenous injection at dose level of 30, 60 and 120 mg/kg once every 2 days from the 6th to 16th day of pregnancy. No significant changes were observed in pregnant rats, embryo and fetal appearance, skeleton and internal organs. The NOAEL of Albuvirtide for the parental pregnant rats, and embryos-fetus development were both 120 mg/kg, i.e. 4 folds of human equivalent dose calculated in terms of body surface area. Toxicokinetic study revealed no significant accumulation of Albuvirtide in pregnant rats.
In the embryo-fetal development toxicity study in rabbit, the pregnant rabbits were given Albuvirtide by intravenous injection at dose levels of 15, 30 and 60 mg/kg once every 3 days from the 6th to 18th day of pregnancy. In the highest dose of 60 mg/kg group, only 1 rabbit (1/13) delivered prematurely, and the percentage of dysostosis of hyoid bone showed an upward trend compared with that of the control group. There were no significant abnormal changes in appearance, bone and internal organs in the other pregnant rabbits and fetal rabbits. The NOAEL of Albuvirtide for the parental rabbits, and embryos-fetus development were all 30 mg/kg, i.e. 3.9 folds of human equivalent dose calculated in terms of exposure. Toxicokinetic study showed no significant accumulation of Albuvirtide in pregnant rabbits.
[Pharmacokinetics]
Pharmacokinetic studies of single dose and multiple dose intravenous administration of Albuvirtide have been conducted in adult subjects with HIV-1 infection. There was a good linear relationship between AUC0-∞ and dose, indicating that Albuvirtide met the linear elimination pattern. The pharmacokinetic parameters of Albuvirtide at a single dose of 320 mg by intravenous infusion were AUC0-∞ 3012.6 ± 373.0 mg•h/L, and Cmax 61.9 ± 5.6 mg/L. In the multiple dose study, the steady pharmacokinetic parameters of Albuvirtide at a dose of 320 mg by intravenous infusion once a week were AUC0-∞ 4946.3 ± 407.1 mg • h/ L, Cmax 57.0 ± 7.9 mg/ L, and Ctrough 6.9 mg/ L.
The distribution and excretion study in rats showed that Albuvirtide was well distributed in all tissues and organs in vivo, and the highest amount of Albuvirtide was present in whole blood, followed by kidney, ovary and other tissues, with the least being in brain, body fat and testes. Albuvirtide was mainly eliminated through kidney in vivo.
In vitro tests demonstrated that Albuvirtide had no significant inhibitory effect on the in vitro activity of six major P450 enzymes (CYP1A2, 2C8, 2C9, 2C19, 2D6 and 3A4) in human liver microsomes.
Special Populations
Gender and race
There are no gender differences in the pharmacokinetics of Albuvirtide, and racial differences in pharmacokinetics have not been established yet.
Children and elderly
The pharmacokinetic study has not yet been conducted in children under 16 years old and elderly above 65 years old.
Hepatic and renal insufficiency
The pharmacokinetic study has not been conducted in patients with hepatic and renal insufficiency.
[Storage]
Preserve in a sealed container under refrigerated (2-8°C) condition, protected from light.
[Package]
20 ml injection vials made of brown low borosilicate glass tubing, 1 vial/carton.
[Shelf-life]
24 months
[Manufacturer]
Company: Frontier Biotechnologies Inc.
Production address: No.7 Building, No. 5 Qiande Road, Hi-Tech Park, Jiangning, Nanjing, China
[Date of Revision of Package Insert]
May 23, 2018
